
Supplementary Material - Computer Methods and Programs in Biomedicine
This website contains supplementary material to the paper:
A. Ramírez-Mena, E. Andrés-León, M. Jesus Alvarez-Cubero, A. Anguita-Ruiz, L.J. Martinez-Gonzalez, J. Alcala-Fdez. Explainable artificial intelligence to predict and identify prostate cancer tissue by gene expression. Computer Methods and Programs in Biomedicine 240 (2023) 107719. doi: 10.1016/j.cmpb.2023.107719
Summary:
- Differential expression analysis
- Datasets
- Final geneset overview
- Machine learning methods considered
- Explainability
- References
Differential expression analysis
After downloading FASTQ files from the TCGA-PRAD project, and obtaining the raw counts for every sample, differential expression analysis was carried out as explained in the paper. Detailed data generated during this processed is available below for download:
| Description | #genes | Download |
|---|---|---|
| DEGs for TCGA-PRAD (full population, satisfying |logFC|>1 and FDR <0.05) | 1991 | |
| DEGs for TCGA-PRAD (paired samples, satisfying |logFC|>1 and FDR <0.05) | 1332 | |
| PRAD-DEGs: intersection of DEGs for TCGA-PRAD from paired and unpaired population (satisfying |logFC|>1 and FDR <0.05) | 1065 | |
| 55TOP-PRAD-DEGs | 55 | |
| PRAD-DEGs-PROST-int-CANCER | 114 | |
| 47-PCa-Genes | 47 |
Datasets
In general, all PC studies of significant size have an imbalance between cases and controls, which is mainly caused by the fact that control biopsies have to be taken from non-tumorous areas of patients with low tumor grade, or are negative biopsies taken from patients with high PSA at the time who may later develop the disease. Healthy individuals never have prostate biopsies taken for research purposes, as this would be ethically unacceptable because it is an invasive technique with side effects. Therefore, the strategy used to select the different populations is state-of-the-art when working with prostate cancer biopsies.
In addition, the TCGA-PRAD population allows raw data to be downloaded after permission has been granted, which was key during the training stage of our algorithm. Other databases, such as GSE183019, which was used as a validation cohort, contains a balanced number of cases and controls, but is smaller, includes only pre-processed data, and lacks ethnic diversity.
The population origin of the data is also a relevant factor to consider, as we wanted to include a wide variety of populations in the study, but no available dataset contained a balanced ethnicity proportion. This is another reason why we adopted the TCGA PRAD repository, which, although unbalanced, provides us with both healthy and prostate cancer-affected tissue samples (some of which are paired), ethnic diversity, and raw RNASEQ data from all samples, which are good conditions for applying our analysis pipeline prior to algorithm training. In fact, despite the complexity we had to manage due to the balanced training dataset, we were able to verify that our algorithm performs similarly in populations with different ancestries, demonstrating its ability to generalize its behavior.
The following table shows the datasets considered in this study. Along with the dataset name, the following fields are shown: the purpose of the dataset for this work TRAINING/VALIDATION; the number of samples in each dataset, distinguising between tumoral samples (T) and controls (NT); observations and a link to the data source.
| Dataset | Purpose | Samples | Observations | Download |
| TCGA-PRAD | Model and parameters training | 498T vs 52NT (52 paired) | Access controlled | |
| GSE22260 | Validation | 20T vs 10NT (10 paired) | ||
| GTEX | Validation | 245NT | ||
| GSE183019 | Validation | 84T vs 84NT | ||
| GSE114740 | Validation | 10T vs 10NT (paired) |
One of the strengths of this system is its ability to classify samples into healthy/tumour samples independently of the genetic ancestry of the individuals in the study. This is an important problem to those systems trained with purely genomic data.
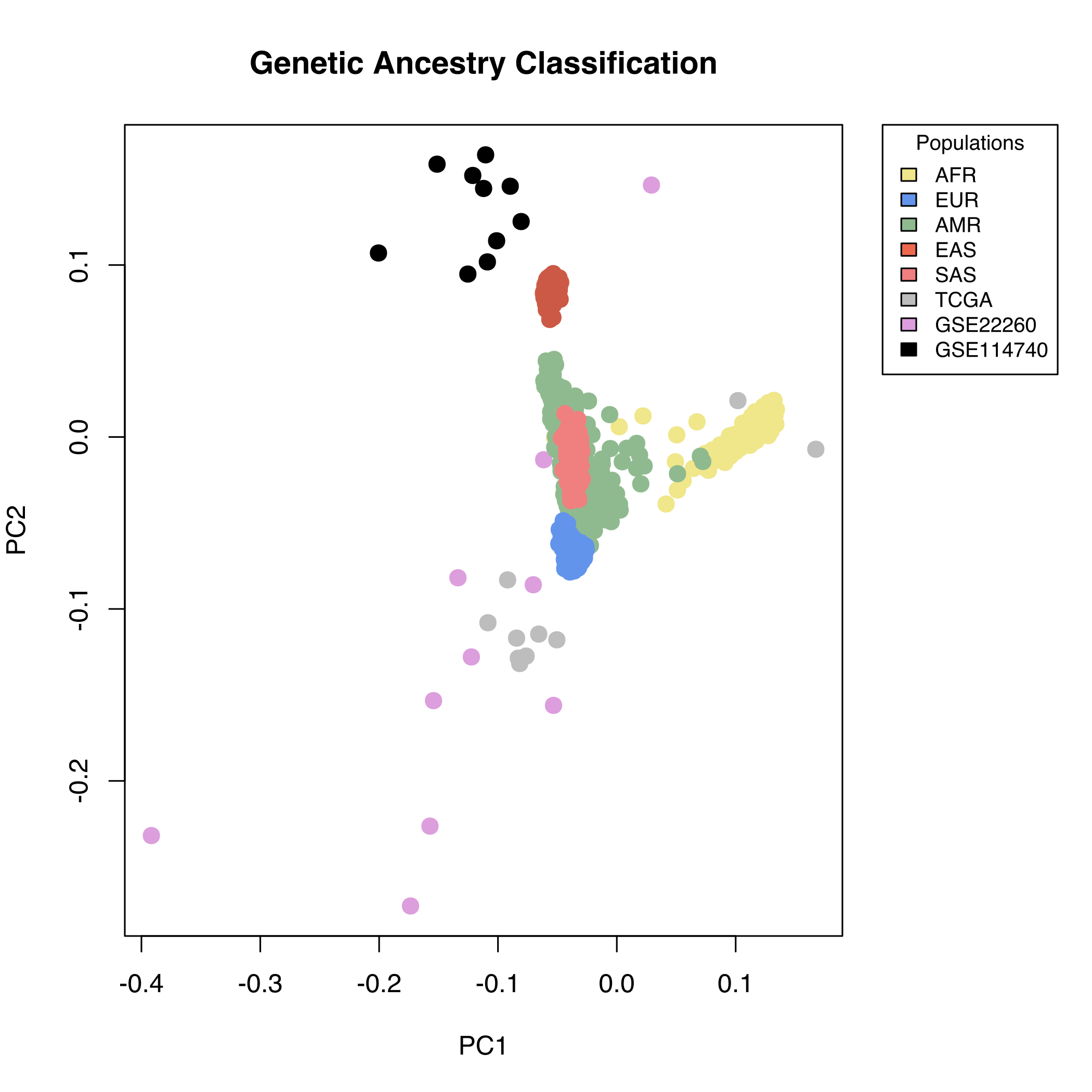
To demonstrate this, non-tumour samples from the GEO datasets: GSE114740, GSE22260 and 10 TCGA samples were clustered, against samples from the 1000 genomes consortium project (1KGP) [1]. In this consortium we found a total of 2504 samples sequenced at a minimum depth of 30x and classified according to their genetic ancestry. No clear information about the population is provided in the GEO datasets. For the TCGA samples, according with the clinical information, 8 white (not Hispanic or Latino) and two black or African American were selected.
To make this clustering more precise, 15,020 polymorphisms known as AIM (ancestry informative markers), have been used, in order to allow a classification into populations [2]. In detail, for each of the non-tumoral samples from the three datasets, fastq files were aligned using BWA [3]. Picard [4] was used to remove duplicate sequences, as well as to sort the reads and index the resulting bam file. These were processed with GATK (Genome Analysis Tool Kit) [5] to identify variants. In the case of the 1000 Genomes data, the variant files provided by the consortium were used. After filtering all the SNPs not related with the AIMs mentioned above with intersectBed (from bedtools) [6], plink was used to cluster the samples based on their genetic ancestry [7].
As shown in Fig. S1, we have 5 populations from 1KGP named AFR (Africans), EUR (Europeans), AMR (Admixed American: Colombians, Peruvians, Mexicans and Puerto Ricans), EAS (East Asians) and SAS (South Asians). From the GSE114740 study, we find that the 10 patients cluster very closely with East Asians, as would be expected given that the samples come from an hospital in Beijing. In the case of the GSE22260 study, the 10 healthy tissue samples cluster close to Europeans as expected given these are samples from North Americans and thus, of European ancestry, with two exception, one sample close to east Asians and another next to AMR/SAS. Similarly, the TCGA samples are from North Americans and therefore also cluster close to Europeans with the exception of the two black or African American samples that cluster close to the African individuals.
Final geneset overview
Below is an brief description of our final geneset 47-PCa-Genes, including their Gene_ID, a brief summary and their location coordinates. The full table, including an extensive description of every gene can be downloaded by clicking the following link:
| Gene_ID | GeneCards_summary | GeneCards_location_coords |
| ACTA2 | This gene encodes one of six different actin proteins. Actins are highly conserved proteins that are involved in cell motility, structure, integrity, and intercellular signaling. The encoded protein is a smooth muscle actin that is involved in vascular contractility and blood pressure homeostasis. Mutations in this gene cause a variety of vascular diseases, such as thoracic aortic disease, coronary artery disease, stroke, and Moyamoya disease, as well as multisystemic smooth muscle dysfunction syndrome. [provided by RefSeq, Sep 2017] /// Actin is a ubiquitous globular protein that is one of the most highly-conserved proteins known. It is found in two main states: G-actin is the globular monomeric form, whereas F-actin forms helical polymers. Both G- and F-actin are intrinsically flexible structures. | chr10:88,935,074-88,991,397 |
| ACTG2 | Actins are highly conserved proteins that are involved in various types of cell motility and in the maintenance of the cytoskeleton. Three types of actins, alpha, beta and gamma, have been identified in vertebrates. Alpha actins are found in muscle tissues and are a major constituent of the contractile apparatus. The beta and gamma actins co-exist in most cell types as components of the cytoskeleton and as mediators of internal cell motility. This gene encodes actin gamma 2; a smooth muscle actin found in enteric tissues. Alternative splicing results in multiple transcript variants encoding distinct isoforms. Based on similarity to peptide cleavage of related actins, the mature protein of this gene is formed by removal of two N-terminal peptides.[provided by RefSeq, Dec 2010] /// Actin is a ubiquitous globular protein that is one of the most highly-conserved proteins known. It is found in two main states: G-actin is the globular monomeric form, whereas F-actin forms helical polymers. Both G- and F-actin are intrinsically flexible structures. | chr2:73,892,314-73,919,865 |
| AMACR | This gene encodes a racemase. The encoded enzyme interconverts pristanoyl-CoA and C27-bile acylCoAs between their (R)- and (S)-stereoisomers. The conversion to the (S)-stereoisomers is necessary for degradation of these substrates by peroxisomal beta-oxidation. Encoded proteins from this locus localize to both mitochondria and peroxisomes. Mutations in this gene may be associated with adult-onset sensorimotor neuropathy, pigmentary retinopathy, and adrenomyeloneuropathy due to defects in bile acid synthesis. Alternatively spliced transcript variants have been described. Read-through transcription also exists between this gene and the upstream neighboring C1QTNF3 (C1q and tumor necrosis factor related protein 3) gene. [provided by RefSeq, Mar 2011] | chr5:33,986,165-34,008,104 |
| ANXA2 | This gene encodes a member of the annexin family. Members of this calcium-dependent phospholipid-binding protein family play a role in the regulation of cellular growth and in signal transduction pathways. This protein functions as an autocrine factor which heightens osteoclast formation and bone resorption. This gene has three pseudogenes located on chromosomes 4, 9 and 10. respectively. Multiple alternatively spliced transcript variants encoding different isoforms have been found for this gene. Annexin A2 expression has been found to correlate with resistance to treatment against various cancer forms. [provided by RefSeq, Dec 2019] | chr15:60.347,134-60.402,883 |
| AOX1 | Aldehyde oxidase produces hydrogen peroxide and, under certain conditions, can catalyze the formation of superoxide. Aldehyde oxidase is a candidate gene for amyotrophic lateral sclerosis. [provided by RefSeq, Jul 2008] | chr2:200.585,952-200.677,064 |
| APOF | The product of this gene is one of the minor apolipoproteins found in plasma. This protein forms complexes with lipoproteins and may be involved in transport and/or esterification of cholesterol. [provided by RefSeq, Jul 2008] /// No disorders were found for APOF Gene. | chr12:56,360.568-56,362,857 |
| BMP5 | This gene encodes a secreted ligand of the TGF-beta (transforming growth factor-beta) superfamily of proteins. Ligands of this family bind various TGF-beta receptors leading to recruitment and activation of SMAD family transcription factors that regulate gene expression. The encoded preproprotein is proteolytically processed to generate each subunit of the disulfide-linked homodimer, which plays a role in bone and cartilage development. Polymorphisms in this gene may be associated with osteoarthritis in human patients. This gene is differentially regulated in multiple human cancers. This gene encodes distinct protein isoforms that may be similarly proteolytically processed. [provided by RefSeq, Jul 2016] | chr6:55,753,653-55,875,590 |
| BMP7 | This gene encodes a secreted ligand of the TGF-beta (transforming growth factor-beta) superfamily of proteins. Ligands of this family bind various TGF-beta receptors leading to recruitment and activation of SMAD family transcription factors that regulate gene expression. The encoded preproprotein is proteolytically processed to generate each subunit of the disulfide-linked homodimer, which plays a role in bone, kidney and brown adipose tissue development. Additionally, this protein induces ectopic bone formation and may promote fracture healing in human patients. [provided by RefSeq, Jul 2016] | chr20:57,168,753-57,266,641 |
| CA14 | Carbonic anhydrases (CAs) are a large family of zinc metalloenzymes that catalyze the reversible hydration of carbon dioxide. They participate in a variety of biological processes, including respiration, calcification, acid-base balance, bone resorption, and the formation of aqueous humor, cerebrospinal fluid, saliva, and gastric acid. They show extensive diversity in tissue distribution and in their subcellular localization. CA XIV is predicted to be a type I membrane protein and shares highest sequence similarity with the other transmembrane CA isoform, CA XII; however, they have different patterns of tissue-specific expression and thus may play different physiologic roles. [provided by RefSeq, Jul 2008] | chr1:150.257,251-150.265,862 |
| CAV1 | The scaffolding protein encoded by this gene is the main component of the caveolae plasma membranes found in most cell types. The protein links integrin subunits to the tyrosine kinase FYN, an initiating step in coupling integrins to the Ras-ERK pathway and promoting cell cycle progression. The gene is a tumor suppressor gene candidate and a negative regulator of the Ras-p42/44 mitogen-activated kinase cascade. Caveolin 1 and caveolin 2 are located next to each other on chromosome 7 and express colocalizing proteins that form a stable hetero-oligomeric complex. Mutations in this gene have been associated with Berardinelli-Seip congenital lipodystrophy. Alternatively spliced transcripts encode alpha and beta isoforms of caveolin 1.[provided by RefSeq, Mar 2010] | chr7:116,524,994-116,561,185 |
| CAV2 | The protein encoded by this gene is a major component of the inner surface of caveolae, small invaginations of the plasma membrane, and is involved in essential cellular functions, including signal transduction, lipid metabolism, cellular growth control and apoptosis. This protein may function as a tumor suppressor. This gene and related family member (CAV1) are located next to each other on chromosome 7, and express colocalizing proteins that form a stable hetero-oligomeric complex. Alternatively spliced transcript variants encoding different isoforms have been identified for this gene. Additional isoforms resulting from the use of alternate in-frame translation initiation codons have also been described, and shown to have preferential localization in the cell (PMID:11238462). [provided by RefSeq, May 2011] | chr7:116,287,380-116,508,541 |
| CNN1 | Predicted to enable actin binding activity. Involved in negative regulation of vascular associated smooth muscle cell proliferation. Located in cytoskeleton. [provided by Alliance of Genome Resources, Nov 2021] | chr19:11,538,767-11,550.323 |
| CXCL13 | B lymphocyte chemoattractant, independently cloned and named Angie, is an antimicrobial peptide and CXC chemokine strongly expressed in the follicles of the spleen, lymph nodes, and Peyer's patches. It preferentially promotes the migration of B lymphocytes (compared to T cells and macrophages), apparently by stimulating calcium influx into, and chemotaxis of, cells expressing Burkitt's lymphoma receptor 1 (BLR-1). It may therefore function in the homing of B lymphocytes to follicles. [provided by RefSeq, Oct 2014] | chr4:77,511,753-77,611,834 |
| CXCR2 | The protein encoded by this gene is a member of the G-protein-coupled receptor family. This protein is a receptor for interleukin 8 (IL8). It binds to IL8 with high affinity, and transduces the signal through a G-protein activated second messenger system. This receptor also binds to chemokine (C-X-C motif) ligand 1 (CXCL1/MGSA), a protein with melanoma growth stimulating activity, and has been shown to be a major component required for serum-dependent melanoma cell growth. This receptor mediates neutrophil migration to sites of inflammation. The angiogenic effects of IL8 in intestinal microvascular endothelial cells are found to be mediated by this receptor. Knockout studies in mice suggested that this receptor controls the positioning of oligodendrocyte precursors in developing spinal cord by arresting their migration. This gene, IL8RA, a gene encoding another high affinity IL8 receptor, as well as IL8RBP, a pseudogene of IL8RB, form a gene cluster in a region mapped to chromosome 2q33-q36. Alternatively spliced variants, encoding the same protein, have been identified. [provided by RefSeq, Nov 2009] /// Chemokine CXC receptors (CXCRs) predominantly recognize CXC chemokines. CXC chemokines are distinguished by having four conserved cysteines, with the first two cysteines being separated by a single amino acid. There are six chemokine CXC receptors. | chr2:218,125,289-218,137,253 |
| DCN | This gene encodes a member of the small leucine-rich proteoglycan family of proteins. Alternative splicing results in multiple transcript variants, at least one of which encodes a preproprotein that is proteolytically processed to generate the mature protein. This protein plays a role in collagen fibril assembly. Binding of this protein to multiple cell surface receptors mediates its role in tumor suppression, including a stimulatory effect on autophagy and inflammation and an inhibitory effect on angiogenesis and tumorigenesis. This gene and the related gene biglycan are thought to be the result of a gene duplication. Mutations in this gene are associated with congenital stromal corneal dystrophy in human patients. [provided by RefSeq, Nov 2015] | chr12:91,140.484-91,183,217 |
| DKK1 | This gene encodes a member of the dickkopf family of proteins. Members of this family are secreted proteins characterized by two cysteine-rich domains that mediate protein-protein interactions. The encoded protein binds to the LRP6 co-receptor and inhibits beta-catenin-dependent Wnt signaling. This gene plays a role in embryonic development and may be important in bone formation in adults. Elevated expression of this gene has been observed in numerous human cancers and this protein may promote proliferation, invasion and growth in cancer cell lines. [provided by RefSeq, Sep 2017] | chr10:52,314,281-52,318,042 |
| DLX1 | This gene encodes a member of a homeobox transcription factor gene family similiar to the Drosophila distal-less gene. The encoded protein is localized to the nucleus where it may function as a transcriptional regulator of signals from multiple TGF-{beta} superfamily members. The encoded protein may play a role in the control of craniofacial patterning and the differentiation and survival of inhibitory neurons in the forebrain. This gene is located in a tail-to-tail configuration with another member of the family on the long arm of chromosome 2. Alternatively spliced transcript variants encoding different isoforms have been described. [provided by RefSeq, Jul 2008] | chr2:172,084,740-172,089,677 |
| DUOX2 | The protein encoded by this gene is a glycoprotein and a member of the NADPH oxidase family. The synthesis of thyroid hormone is catalyzed by a protein complex located at the apical membrane of thyroid follicular cells. This complex contains an iodide transporter, thyroperoxidase, and a peroxide generating system that includes this encoded protein and DUOX1. This protein is known as dual oxidase because it has both a peroxidase homology domain and a gp91phox domain. [provided by RefSeq, Jul 2008] | chr15:45,092,650-45,114,172 |
| DUOXA2 | This gene encodes an endoplasmic reticulum protein that is necessary for proper cellular localization and maturation of functional dual oxidase 2. Mutations in this gene have been associated with thyroid dyshormonogenesis 5.[provided by RefSeq, Feb 2010] | chr15:45,114,321-45,118,421 |
| EPHA2 | This gene belongs to the ephrin receptor subfamily of the protein-tyrosine kinase family. EPH and EPH-related receptors have been implicated in mediating developmental events, particularly in the nervous system. Receptors in the EPH subfamily typically have a single kinase domain and an extracellular region containing a Cys-rich domain and 2 fibronectin type III repeats. The ephrin receptors are divided into 2 groups based on the similarity of their extracellular domain sequences and their affinities for binding ephrin-A and ephrin-B ligands. This gene encodes a protein that binds ephrin-A ligands. Mutations in this gene are the cause of certain genetically-related cataract disorders.[provided by RefSeq, May 2010] /// Eph receptors are the largest family of receptor tyrosine kinases (RTKs) and are divided into two subclasses, EphA and EphB. Originally identified as mediators of axon guidance, Eph receptors are implicated in many processes, particularly cancer development and progression. | chr1:16,124,337-16,156,104 |
| ERG | This gene encodes a member of the erythroblast transformation-specific (ETS) family of transcriptions factors. All members of this family are key regulators of embryonic development, cell proliferation, differentiation, angiogenesis, inflammation, and apoptosis. The protein encoded by this gene is mainly expressed in the nucleus. It contains an ETS DNA-binding domain and a PNT (pointed) domain which is implicated in the self-association of chimeric oncoproteins. This protein is required for platelet adhesion to the subendothelium, inducing vascular cell remodeling. It also regulates hematopoesis, and the differentiation and maturation of megakaryocytic cells. This gene is involved in chromosomal translocations, resulting in different fusion gene products, such as TMPSSR2-ERG and NDRG1-ERG in prostate cancer, EWS-ERG in Ewing's sarcoma and FUS-ERG in acute myeloid leukemia. More than two dozens of transcript variants generated from combinatorial usage of three alternative promoters and multiple alternative splicing events have been reported, but the full-length nature of many of these variants has not been determined. [provided by RefSeq, Apr 2014] | chr21:38,367,261-38,661,783 |
| FGFR2 | The protein encoded by this gene is a member of the fibroblast growth factor receptor family, where amino acid sequence is highly conserved between members and throughout evolution. FGFR family members differ from one another in their ligand affinities and tissue distribution. A full-length representative protein consists of an extracellular region, composed of three immunoglobulin-like domains, a single hydrophobic membrane-spanning segment and a cytoplasmic tyrosine kinase domain. The extracellular portion of the protein interacts with fibroblast growth factors, setting in motion a cascade of downstream signals, ultimately influencing mitogenesis and differentiation. This particular family member is a high-affinity receptor for acidic, basic and/or keratinocyte growth factor, depending on the isoform. Mutations in this gene are associated with Crouzon syndrome, Pfeiffer syndrome, Craniosynostosis, Apert syndrome, Jackson-Weiss syndrome, Beare-Stevenson cutis gyrata syndrome, Saethre-Chotzen syndrome, and syndromic craniosynostosis. Multiple alternatively spliced transcript variants encoding different isoforms have been noted for this gene. [provided by RefSeq, Jan 2009] /// The FGFR proteins are involved in a wide array of pathways known to play a signficant role in cancer. Activation of these receptors can lead to activation of the RAS-MAPK pathway and the PI3K-AKT pathway, among others. The mechanisms by which FGFR can be misregulated vary between cancers. Amplification of the receptors has been observed in lung and breast cancers, coding mutations and deletions have been seen in many cancers, and more recently, FGFR fusions that lead to pathway actiation have been demonstrated to have oncogenic potential across multiple cancer types. The targeted therapeutics ponatinib, dovitinib and pazopanib have seen success in treating over-active FGFR signalling, prompting use of diagnostic sequencing targeting the FGFR genes, especially in lung cancer patients. /// Fibroblast growth factors (FGFs) are mitogenic signaling molecules that have roles in angiogenesis, wound healing, cell migration, neural outgrowth and embryonic development. FGF receptors (FGFRs) are transmembrane catalytic receptors with intracellular tyrosine kinase activity. | chr10:121,478,330-121,598,458 |
| FLNA | The protein encoded by this gene is an actin-binding protein that crosslinks actin filaments and links actin filaments to membrane glycoproteins. The encoded protein is involved in remodeling the cytoskeleton to effect changes in cell shape and migration. This protein interacts with integrins, transmembrane receptor complexes, and second messengers. Defects in this gene are a cause of several syndromes, including periventricular nodular heterotopias (PVNH1, PVNH4), otopalatodigital syndromes (OPD1, OPD2), frontometaphyseal dysplasia (FMD), Melnick-Needles syndrome (MNS), and X-linked congenital idiopathic intestinal pseudoobstruction (CIIPX). Two transcript variants encoding different isoforms have been found for this gene.[provided by RefSeq, Mar 2009] | chrX:154,348,524-154,374,634 |
| GLYATL1 | Enables glutamine N-acyltransferase activity. Involved in glutamine metabolic process. Predicted to be located in mitochondrion. [provided by Alliance of Genome Resources, Nov 2021] | chr11:58,905,398-59,043,527 |
| HOXC6 | This gene belongs to the homeobox family, members of which encode a highly conserved family of transcription factors that play an important role in morphogenesis in all multicellular organisms. Mammals possess four similar homeobox gene clusters, HOXA, HOXB, HOXC and HOXD, which are located on different chromosomes and consist of 9 to 11 genes arranged in tandem. This gene, HOXC6, is one of several HOXC genes located in a cluster on chromosome 12. Three genes, HOXC5, HOXC4 and HOXC6, share a 5' non-coding exon. Transcripts may include the shared exon spliced to the gene-specific exons, or they may include only the gene-specific exons. Alternatively spliced transcript variants encoding different isoforms have been identified for HOXC6. Transcript variant two includes the shared exon, and transcript variant one includes only gene-specific exons. [provided by RefSeq, Jul 2008] | chr12:53,990.624-54,030.823 |
| HPN | This gene encodes a type II transmembrane serine protease that may be involved in diverse cellular functions, including blood coagulation and the maintenance of cell morphology. Expression of the encoded protein is associated with the growth and progression of cancers, particularly prostate cancer. The protein is cleaved into a catalytic serine protease chain and a non-catalytic scavenger receptor cysteine-rich chain, which associate via a single disulfide bond. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Jan 2013] | chr19:35,040.506-35,066,573 |
| KCNMA1 | MaxiK channels are large conductance, voltage and calcium-sensitive potassium channels which are fundamental to the control of smooth muscle tone and neuronal excitability. MaxiK channels can be formed by 2 subunits: the pore-forming alpha subunit, which is the product of this gene, and the modulatory beta subunit. Intracellular calcium regulates the physical association between the alpha and beta subunits. Alternatively spliced transcript variants encoding different isoforms have been identified. [provided by RefSeq, Jul 2008] /// Calcium (Ca2+) -activated potassium channels (KCa) are a group of 6/7-TM ion channels that selectively transport K+ ions across biological membranes. They are broadly classified into three subtypes: SK, IK and BK channels (small, intermediate and big conductance). | chr10:76,869,601-77,638,369 |
| KRT7 | The protein encoded by this gene is a member of the keratin gene family. The type II cytokeratins consist of basic or neutral proteins which are arranged in pairs of heterotypic keratin chains coexpressed during differentiation of simple and stratified epithelial tissues. This type II cytokeratin is specifically expressed in the simple epithelia lining the cavities of the internal organs and in the gland ducts and blood vessels. The genes encoding the type II cytokeratins are clustered in a region of chromosome 12q12-q13. Alternative splicing may result in several transcript variants; however, not all variants have been fully described. [provided by RefSeq, Jul 2008] | chr12:52,232,520-52,252,667 |
| LMO3 | The protein encoded by this gene belongs to the rhombotin family of cysteine-rich LIM domain oncogenes. This gene is predominantly expressed in the brain. Related family members, LMO1 and LMO2 on chromosome 11, have been reported to be involved in chromosomal translocations in T-cell leukemia. Many alternatively spliced transcript variants have been found for this gene. [provided by RefSeq, Aug 2011] | chr12:16,548,372-16,610.594 |
| LMOD1 | The leiomodin 1 protein has a putative membrane-spanning region and 2 types of tandemly repeated blocks. The transcript is expressed in all tissues tested, with the highest levels in thyroid, eye muscle, skeletal muscle, and ovary. Increased expression of leiomodin 1 may be linked to Graves' disease and thyroid-associated ophthalmopathy. [provided by RefSeq, Jul 2008] | chr1:201,896,456-201,946,588 |
| MMP9 | Proteins of the matrix metalloproteinase (MMP) family are involved in the breakdown of extracellular matrix in normal physiological processes, such as embryonic development, reproduction, and tissue remodeling, as well as in disease processes, such as arthritis and metastasis. Most MMP's are secreted as inactive proproteins which are activated when cleaved by extracellular proteinases. The enzyme encoded by this gene degrades type IV and V collagens. Studies in rhesus monkeys suggest that the enzyme is involved in IL-8-induced mobilization of hematopoietic progenitor cells from bone marrow, and murine studies suggest a role in tumor-associated tissue remodeling. [provided by RefSeq, Jul 2008] /// Matrix metalloproteases (matrix metalloproteinase, MMPs), also called matrixins, are zinc-dependent endopeptidases and the major proteases in ECM degradation. MMPs are capable of degrading several extracellular molecules and a number of bioactive molecules. | chr20:46,008,908-46,016,561 |
| MYL9 | Myosin, a structural component of muscle, consists of two heavy chains and four light chains. The protein encoded by this gene is a myosin light chain that may regulate muscle contraction by modulating the ATPase activity of myosin heads. The encoded protein binds calcium and is activated by myosin light chain kinase. Two transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Jul 2008] /// Myosins are a large family of motor proteins that share the common features of ATP hydrolysis (ATPase enzyme activity), actin binding and potential for kinetic energy transduction. Originally isolated from muscle cells, almost all eukaryotic cells are known to contain myosins. | chr20:36,541,497-36,551,447 |
| MYLK | This gene, a muscle member of the immunoglobulin gene superfamily, encodes myosin light chain kinase which is a calcium/calmodulin dependent enzyme. This kinase phosphorylates myosin regulatory light chains to facilitate myosin interaction with actin filaments to produce contractile activity. This gene encodes both smooth muscle and nonmuscle isoforms. In addition, using a separate promoter in an intron in the 3' region, it encodes telokin, a small protein identical in sequence to the C-terminus of myosin light chain kinase, that is independently expressed in smooth muscle and functions to stabilize unphosphorylated myosin filaments. A pseudogene is located on the p arm of chromosome 3. Four transcript variants that produce four isoforms of the calcium/calmodulin dependent enzyme have been identified as well as two transcripts that produce two isoforms of telokin. Additional variants have been identified but lack full length transcripts. [provided by RefSeq, Jul 2008] /// Myosin Light Chain Kinases (MLCKs) are a group of protein serine/threonine kinases that are currently divided into two subtypes. MLCK1 is found in smooth muscle and phosphorylates myosin II regulatory light chains at Ser19. MLCK2 is located in the striated muscle. | chr3:123,610.049-123,884,332 |
| MYO6 | This gene encodes a reverse-direction motor protein that moves toward the minus end of actin filaments and plays a role in intracellular vesicle and organelle transport. The protein consists of a motor domain containing an ATP- and an actin-binding site and a globular tail which interacts with other proteins. This protein maintains the structural integrity of inner ear hair cells and mutations in this gene cause non-syndromic autosomal dominant and recessive hearing loss. Alternative splicing results in multiple transcript variants encoding distinct isoforms. [provided by RefSeq, Jul 2014] | chr6:75,749,201-75,919,537 |
| MYOCD | This gene encodes a nuclear protein, which is expressed in heart, aorta, and in smooth muscle cell-containing tissues. It functions as a transcriptional co-activator of serum response factor (SRF) and modulates expression of cardiac and smooth muscle-specific SRF-target genes, and thus may play a crucial role in cardiogenesis and differentiation of the smooth muscle cell lineage. Alternatively spliced transcript variants encoding different isoforms have been found for this gene.[provided by RefSeq, Sep 2009] | chr17:12,665,890-12,768,949 |
| OR51E2 | Olfactory receptors interact with odorant molecules in the nose, to initiate a neuronal response that triggers the perception of a smell. The olfactory receptor proteins are members of a large family of G-protein-coupled receptors (GPCR) arising from single coding-exon genes. Olfactory receptors share a 7-transmembrane domain structure with many neurotransmitter and hormone receptors and are responsible for the recognition and G protein-mediated transduction of odorant signals. The olfactory receptor gene family is the largest in the genome. The nomenclature assigned to the olfactory receptor genes and proteins for this organism is independent of other organisms. [provided by RefSeq, Jul 2008] | chr11:4,680.171-4,697,854 |
| PLA2G7 | The protein encoded by this gene is a secreted enzyme that catalyzes the degradation of platelet-activating factor to biologically inactive products. Defects in this gene are a cause of platelet-activating factor acetylhydrolase deficiency. Two transcript variants encoding the same protein have been found for this gene.[provided by RefSeq, Dec 2009] /// Phospholipases are a group of enzymes that hydrolyze phospholipids into fatty acids and other lipophilic molecules. There are four major classes; phospholipase A, phospholipase B, phosphoinositide-specific phospholipase C and phospholipase D. | chr6:46,700.558-46,735,836 |
| PTGS1 | This is one of two genes encoding similar enzymes that catalyze the conversion of arachidonate to prostaglandin. The encoded protein regulates angiogenesis in endothelial cells, and is inhibited by nonsteroidal anti-inflammatory drugs such as aspirin. Based on its ability to function as both a cyclooxygenase and as a peroxidase, the encoded protein has been identified as a moonlighting protein. The protein may promote cell proliferation during tumor progression. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Nov 2021] /// Cyclooxygenase (also known as COX, Prostaglandin-endoperoxide synthase, Prostaglandin G/H synthase) is expressed in cells in three isoforms. COX-1 (constitutive) and COX-2 (inducible) isoforms catalyze the rate-limiting step of prostaglandin production. | chr9:122,369,906-122,395,703 |
| RNF112 | This gene encodes a member of the RING finger protein family of transcription factors. The protein is primarily expressed in brain. The gene is located within the Smith-Magenis syndrome region on chromosome 17. [provided by RefSeq, Jul 2008] | chr17:19,406,979-19,417,276 |
| SLPI | This gene encodes a secreted inhibitor which protects epithelial tissues from serine proteases. It is found in various secretions including seminal plasma, cervical mucus, and bronchial secretions, and has affinity for trypsin, leukocyte elastase, and cathepsin G. Its inhibitory effect contributes to the immune response by protecting epithelial surfaces from attack by endogenous proteolytic enzymes. This antimicrobial protein has antibacterial, antifungal and antiviral activity. [provided by RefSeq, Nov 2014] | chr20:45,252,239-45,254,564 |
| SORBS1 | This gene encodes a CBL-associated protein which functions in the signaling and stimulation of insulin. Mutations in this gene may be associated with human disorders of insulin resistance. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Mar 2014] | chr10:95,311,771-95,561,439 |
| SRARP | Enables estrogen receptor binding activity. Involved in positive regulation of intracellular estrogen receptor signaling pathway. Located in cytoplasm and nucleus. [provided by Alliance of Genome Resources, Nov 2021] | chr1:16,004,236-16,008,807 |
| SVIL | This gene encodes a bipartite protein with distinct amino- and carboxy-terminal domains. The amino-terminus contains nuclear localization signals and the carboxy-terminus contains numerous consecutive sequences with extensive similarity to proteins in the gelsolin family of actin-binding proteins, which cap, nucleate, and/or sever actin filaments. The gene product is tightly associated with both actin filaments and plasma membranes, suggesting a role as a high-affinity link between the actin cytoskeleton and the membrane. The encoded protein appears to aid in both myosin II assembly during cell spreading and disassembly of focal adhesions. Several transcript variants encoding different isoforms of supervillin have been described. [provided by RefSeq, Apr 2016] | chr10:29,457,338-29,736,959 |
| TDRD1 | This gene encodes a protein containing a tudor domain that is thought to function in the suppression of transposable elements during spermatogenesis. The related protein in mouse forms a complex with piRNAs and Piwi proteins to promote methylation and silencing of target sequences. This gene was observed to be upregulated by ETS transcription factor ERG in prostate tumors. [provided by RefSeq, Sep 2018] | chr10:114,174,442-114,232,669 |
| TFF3 | Members of the trefoil family are characterized by having at least one copy of the trefoil motif, a 40-amino acid domain that contains three conserved disulfides. They are stable secretory proteins expressed in gastrointestinal mucosa. Their functions are not defined, but they may protect the mucosa from insults, stabilize the mucus layer and affect healing of the epithelium. This gene is expressed in goblet cells of the intestines and colon. This gene and two other related trefoil family member genes are found in a cluster on chromosome 21. [provided by RefSeq, Jul 2008] | chr21:42,311,667-42,315,409 |
| TGFBR3 | This locus encodes the transforming growth factor (TGF)-beta type III receptor. The encoded receptor is a membrane proteoglycan that often functions as a co-receptor with other TGF-beta receptor superfamily members. Ectodomain shedding produces soluble TGFBR3, which may inhibit TGFB signaling. Decreased expression of this receptor has been observed in various cancers. Alternatively spliced transcript variants encoding different isoforms have been identified for this gene.[provided by RefSeq, Sep 2010] | chr1:91,680.343-91,906,335 |
| TIMP3 | This gene belongs to the TIMP gene family. The proteins encoded by this gene family are inhibitors of the matrix metalloproteinases, a group of peptidases involved in degradation of the extracellular matrix (ECM). Expression of this gene is induced in response to mitogenic stimulation and this netrin domain-containing protein is localized to the ECM. Mutations in this gene have been associated with the autosomal dominant disorder Sorsby's fundus dystrophy. [provided by RefSeq, Jul 2008] | chr22:32,801,705-32,863,041 |
Table S1. Brief description of the 47-PCa-Genes geneset, including Gene_ID, summary and location.
Machine learning methods considered
A brief description of every ML method considered in this work is shown below, including a short explanation of the parameteres trained for each one:
- Random forest [8]: This is a supervised machine learning algorithm which can be used both for regression and classification. It works by building a forest of trees with different samples and attributes and its output is based on the majority vote of those trees (for classification) or their average result (for regression).
The "mtry" parameter for this method has been tuned by running a stratified 5-fold cross-validation.This parameter controls the number of variables randomly sampled as candidates at each tree split. - XGBoost [9]: This is a powerful machine learning algorithm that combines weak predictive models, typically decision trees, to create a stronger and more accurate model. It uses a gradient boosting framework, iteratively improving the model by minimizing the residual errors. XGBoost is known for its scalability, speed, and performance in various tasks such as classification, regression, and ranking. It incorporates regularization techniques to prevent overfitting and provides interpretability features, like feature importance, to gain insights from the data. The following parameters were tuned:
- nrounds: The number of boosting rounds or decision trees to build.
- eta: The learning rate, controlling the contribution of each tree.
- max_depth: The maximum depth of each decision tree in the ensemble.
- subsample: The fraction of observations to randomly sample for each tree.
- colsample_bytree: The fraction of features to randomly sample for each tree.
- min_child_weight: The minimum sum of instance weight needed in a child for further partition.
- gamma: The minimum loss reduction required to make a further partition on a leaf node.
- KNN [10]: This is a supervised machine learning algorithm which can be used both for regression and classification. It works by computing the distance between a query and all the samples present in the data, selecting then the k nearest instances to the query and averaging their labels (for regression) or voting for the most frequent label (if it is a classification problem).
The "k" parameter for this method has been tuned by running a stratified 5-fold cross-validation. - rpart [11]: This is a supervised machine learning algorithm which can be used both for regression and classification. The rpart library, available for R, implements recursive partitioning and is used to build classification and regression trees (CART). According to their creators, it is an implementation of most of the functionality of the 1984 book by Breiman, Friedman, Olhsen and Stone. A cart model's output is a decision tree, where each node represents a prediction for the outcome variable and each fork is a split based on a predictor variable.
The "complexity parameter" (CP) argument for this method has been tuned by running a stratified 5-fold cross-validation. The role of this parameter is to prune any split that does not improve the fit by, at least CP.
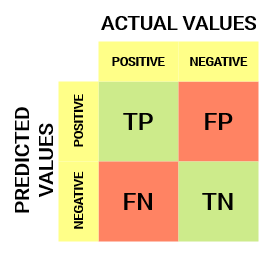
As described in the article, a stratified 5-fold CV with 5 repeats was used for each method and data balancing strategy during the training stage. Below, the results for every iteration for each method are displayed (only the best class/balancing strategy for each method is shown). Given the confusion matrix shown in Fig. S2 for a binary classification problem with two classes (positive and negative), where True Positives (TP; an example in which the model correctly predicted the positive class), False Positives (FP; an example in which the model mistakenly predicted the positive class), False Negatives (FN; an example in which the model mistakenly predicted the negative class) and True Negatives (TN; an example in which the model correctly predicted the negative class) are represented, we describe the metrics considered in this study.
- G-mean: This metric provides a combination of sensitivity and specificity by computing their geometric mean.
- Sensitivity: Measures what proportion of actual positives are correctly predicted as positive. Sensitivity = (True Positive)/(True Positive + False Negative)
- Specificity: This measure is defined as the proportion of actual negatives, which got predicted as the negative class. Specificity = (True Negative)/(True Negative + False Positive)
- AUC: Calculates the area under a Receiver Operator Characteristic(ROC) curve for a classifier. A ROC plots True Positive Rate (TPR) agains False Positive Rate (FPR) at various threshold values. AUC measures the area under this curve.
- F1: F-score, also called the F1-score, is a measure of a model’s accuracy on a dataset. It is used to evaluate binary classification systems, which classify examples into ‘positive’ or ‘negative’. The F-score is a way of combining the precision and recall of the model, and it is defined as the harmonic mean of the model’s precision and recall.
Note that RF and XGBoost are two ensemble learning methods that aim to improve prediction accuracy by combining the results of several simpler decision tree models. Both models can handle high-dimensional data, are robust to outliers and noise, and use regularization techniques to control model complexity and prevent overfitting. However, both models provided statistically similar results in the experimental study. For the sake of clarity for the reader, we decided to include only the results obtained by RF in our work because it obtained slightly better average results in 3 of the 5 quality measures (G-mean, specificity, and F1) used in our analysis.
The following table shows the mean across the 25 test sets for each balancing strategy in the test sets during the experimental analysis.
| Method | Class Bal. | G-mean | Sens. | Spec. | AUC | F1 |
| RF | Undersampling | 0.91 | 0.90 | 0.92 | 0.95 | 0.69 |
| RF | Hybrid | 0.90 | 0.85 | 0.95 | 0.96 | 0.74 |
| RF | Upsampling | 0.84 | 0.71 | 0.99 | 0.96 | 0.76 |
| RF | Weight | 0.80 | 0.65 | 0.99 | 0.96 | 0.73 |
| RF | - | 0.79 | 0.63 | 0.99 | 0.96 | 0.71 |
| XGBoost | Undersampling | 0.90 | 0.91 | 0.90 | 0.95 | 0.65 |
| XGBoost | Hybrid | 0.90 | 0.87 | 0.94 | 0.96 | 0.72 |
| XGBoost | Upsampling | 0.88 | 0.81 | 0.97 | 0.96 | 0.80 |
| XGBoost | Weight | 0.00 | 1.00 | 0.00 | 0.96 | 0.75 |
| XGBoost | - | 0.82 | 0.70 | 0.98 | 0.96 | 0.74 |
| KNN | Undersampling | 0.89 | 0.92 | 0.86 | 0.94 | 0.58 |
| KNN | Hybrid | 0.89 | 0.91 | 0.88 | 0.93 | 0.61 |
| KNN | Upsampling | 0.88 | 0.92 | 0.84 | 0.93 | 0.54 |
| KNN | Weight | 0.70 | 0.50 | 0.99 | 0.93 | 0.60 |
| KNN | - | 0.70 | 0.50 | 0.99 | 0.92 | 0.60 |
| rpart | Undersampling | 0.87 | 0.85 | 0.88 | 0.87 | 0.57 |
| rpart | Hybrid | 0.86 | 0.82 | 0.89 | 0.86 | 0.58 |
| rpart | Upsampling | 0.85 | 0.83 | 0.88 | 0.85 | 0.55 |
| rpart | Weight | 0.85 | 0.82 | 0.88 | 0.85 | 0.56 |
| rpart | - | 0.73 | 0.56 | 0.97 | 0.77 | 0.59 |
Below are several tables with the results for each iteration and repeat during the training stage on the discovery population (TCGA-PRAD) for each algorithm considered with the class balancing strategy that worked best for each one.
RF (undersampling strategy)
| Iteration | G-mean | Sensitivity | Specificity | AUC | F1 |
| Fold1.Rep1 | 0.86 | 0.8 | 0.92 | 0.88 | 0.62 |
| Fold1.Rep2 | 0.93 | 0.91 | 0.95 | 0.96 | 0.77 |
| Fold1.Rep3 | 0.84 | 0.73 | 0.96 | 0.9 | 0.7 |
| Fold1.Rep4 | 0.85 | 0.8 | 0.91 | 0.91 | 0.59 |
| Fold1.Rep5 | 0.95 | 1 | 0.91 | 0.99 | 0.69 |
| Fold2.Rep1 | 0.9 | 0.91 | 0.89 | 0.99 | 0.62 |
| Fold2.Rep2 | 0.88 | 0.9 | 0.87 | 0.9 | 0.56 |
| Fold2.Rep3 | 0.96 | 1 | 0.93 | 0.99 | 0.74 |
| Fold2.Rep4 | 0.89 | 0.82 | 0.96 | 0.91 | 0.75 |
| Fold2.Rep5 | 0.78 | 0.64 | 0.95 | 0.9 | 0.61 |
| Fold3.Rep1 | 0.92 | 0.9 | 0.94 | 0.9 | 0.72 |
| Fold3.Rep2 | 0.95 | 1 | 0.9 | 0.98 | 0.67 |
| Fold3.Rep3 | 0.98 | 1 | 0.97 | 0.99 | 0.88 |
| Fold3.Rep4 | 0.96 | 1 | 0.92 | 0.97 | 0.71 |
| Fold3.Rep5 | 0.83 | 0.8 | 0.86 | 0.92 | 0.5 |
| Fold4.Rep1 | 0.88 | 0.9 | 0.86 | 0.98 | 0.55 |
| Fold4.Rep2 | 0.92 | 0.91 | 0.94 | 0.95 | 0.74 |
| Fold4.Rep3 | 0.86 | 0.9 | 0.82 | 0.95 | 0.49 |
| Fold4.Rep4 | 0.99 | 1 | 0.98 | 1 | 0.92 |
| Fold4.Rep5 | 0.98 | 1 | 0.96 | 1 | 0.83 |
| Fold5.Rep1 | 0.97 | 1 | 0.94 | 0.98 | 0.79 |
| Fold5.Rep2 | 0.98 | 1 | 0.97 | 0.99 | 0.87 |
| Fold5.Rep3 | 0.91 | 1 | 0.83 | 0.99 | 0.54 |
| Fold5.Rep4 | 0.86 | 0.8 | 0.93 | 0.93 | 0.64 |
| Fold5.Rep5 | 0.91 | 0.91 | 0.91 | 0.97 | 0.67 |
Table S2. Detailed results for each iteration in the training stage (5-fold CV with 5 repeats) for RF (undersampling strategy) over the discovery population.
KNN (hybrid strategy)
| Iteration | G-mean | Sensitivity | Specificity | AUC | F1 |
| Fold1.Rep1 | 0.86 | 0.91 | 0.81 | 0.95 | 0.5 |
| Fold1.Rep2 | 0.86 | 0.8 | 0.93 | 0.95 | 0.64 |
| Fold1.Rep3 | 0.91 | 0.91 | 0.92 | 0.95 | 0.69 |
| Fold1.Rep4 | 0.88 | 0.91 | 0.86 | 0.93 | 0.57 |
| Fold1.Rep5 | 0.89 | 0.9 | 0.88 | 0.95 | 0.58 |
| Fold2.Rep1 | 0.94 | 1 | 0.88 | 0.96 | 0.62 |
| Fold2.Rep2 | 0.84 | 0.82 | 0.87 | 0.91 | 0.55 |
| Fold2.Rep3 | 0.89 | 0.9 | 0.89 | 0.98 | 0.6 |
| Fold2.Rep4 | 0.92 | 0.9 | 0.94 | 0.92 | 0.72 |
| Fold2.Rep5 | 0.89 | 0.9 | 0.89 | 0.89 | 0.6 |
| Fold3.Rep1 | 0.95 | 1 | 0.91 | 0.97 | 0.69 |
| Fold3.Rep2 | 0.94 | 1 | 0.88 | 0.96 | 0.62 |
| Fold3.Rep3 | 0.8 | 0.73 | 0.89 | 0.84 | 0.53 |
| Fold3.Rep4 | 0.94 | 1 | 0.89 | 0.98 | 0.67 |
| Fold3.Rep5 | 0.94 | 1 | 0.89 | 0.97 | 0.65 |
| Fold4.Rep1 | 0.79 | 0.73 | 0.85 | 0.88 | 0.47 |
| Fold4.Rep2 | 0.95 | 1 | 0.9 | 0.98 | 0.67 |
| Fold4.Rep3 | 0.9 | 1 | 0.81 | 0.93 | 0.51 |
| Fold4.Rep4 | 0.85 | 0.8 | 0.9 | 0.92 | 0.57 |
| Fold4.Rep5 | 0.87 | 0.91 | 0.83 | 0.88 | 0.53 |
| Fold5.Rep1 | 0.91 | 0.9 | 0.93 | 0.93 | 0.69 |
| Fold5.Rep2 | 0.89 | 0.91 | 0.87 | 0.89 | 0.59 |
| Fold5.Rep3 | 0.95 | 1 | 0.91 | 0.97 | 0.69 |
| Fold5.Rep4 | 0.84 | 0.8 | 0.89 | 0.9 | 0.55 |
| Fold5.Rep5 | 0.91 | 0.91 | 0.91 | 0.92 | 0.67 |
Table S3. Detailed results for each iteration in the training stage (5-fold CV with 5 repeats) for KNN (hybrid strategy) over the discovery population.
rpart (undersampling strategy)
| Iteration | G-mean | Sensitivity | Specificity | AUC | F1 |
| Fold1.Rep1 | 0.95 | 1 | 0.9 | 0.95 | 0.69 |
| Fold1.Rep2 | 0.85 | 0.9 | 0.81 | 0.86 | 0.47 |
| Fold1.Rep3 | 0.89 | 0.91 | 0.87 | 0.89 | 0.59 |
| Fold1.Rep4 | 0.96 | 1 | 0.92 | 0.96 | 0.73 |
| Fold1.Rep5 | 0.84 | 0.82 | 0.87 | 0.84 | 0.55 |
| Fold2.Rep1 | 0.84 | 0.8 | 0.88 | 0.84 | 0.53 |
| Fold2.Rep2 | 0.81 | 0.73 | 0.9 | 0.81 | 0.55 |
| Fold2.Rep3 | 0.83 | 0.8 | 0.87 | 0.84 | 0.52 |
| Fold2.Rep4 | 0.84 | 0.8 | 0.88 | 0.84 | 0.53 |
| Fold2.Rep5 | 0.85 | 0.91 | 0.79 | 0.85 | 0.48 |
| Fold3.Rep1 | 0.75 | 0.6 | 0.93 | 0.77 | 0.52 |
| Fold3.Rep2 | 0.9 | 0.91 | 0.89 | 0.9 | 0.62 |
| Fold3.Rep3 | 0.8 | 0.73 | 0.89 | 0.81 | 0.53 |
| Fold3.Rep4 | 0.92 | 1 | 0.85 | 0.92 | 0.59 |
| Fold3.Rep5 | 0.85 | 1 | 0.73 | 0.86 | 0.43 |
| Fold4.Rep1 | 0.91 | 0.91 | 0.92 | 0.91 | 0.69 |
| Fold4.Rep2 | 0.9 | 0.9 | 0.91 | 0.9 | 0.64 |
| Fold4.Rep3 | 0.8 | 0.8 | 0.81 | 0.8 | 0.43 |
| Fold4.Rep4 | 0.86 | 0.9 | 0.83 | 0.86 | 0.5 |
| Fold4.Rep5 | 0.81 | 0.7 | 0.93 | 0.81 | 0.58 |
| Fold5.Rep1 | 0.85 | 0.8 | 0.9 | 0.85 | 0.57 |
| Fold5.Rep2 | 0.87 | 0.8 | 0.94 | 0.87 | 0.67 |
| Fold5.Rep3 | 0.92 | 0.9 | 0.95 | 0.92 | 0.75 |
| Fold5.Rep4 | 0.83 | 0.8 | 0.87 | 0.83 | 0.52 |
| Fold5.Rep5 | 0.91 | 0.9 | 0.92 | 0.91 | 0.67 |
Table S4. Detailed results for each iteration in the training stage (5-fold CV with 5 repeats) for rpart (undersampling strategy) over the discovery population.
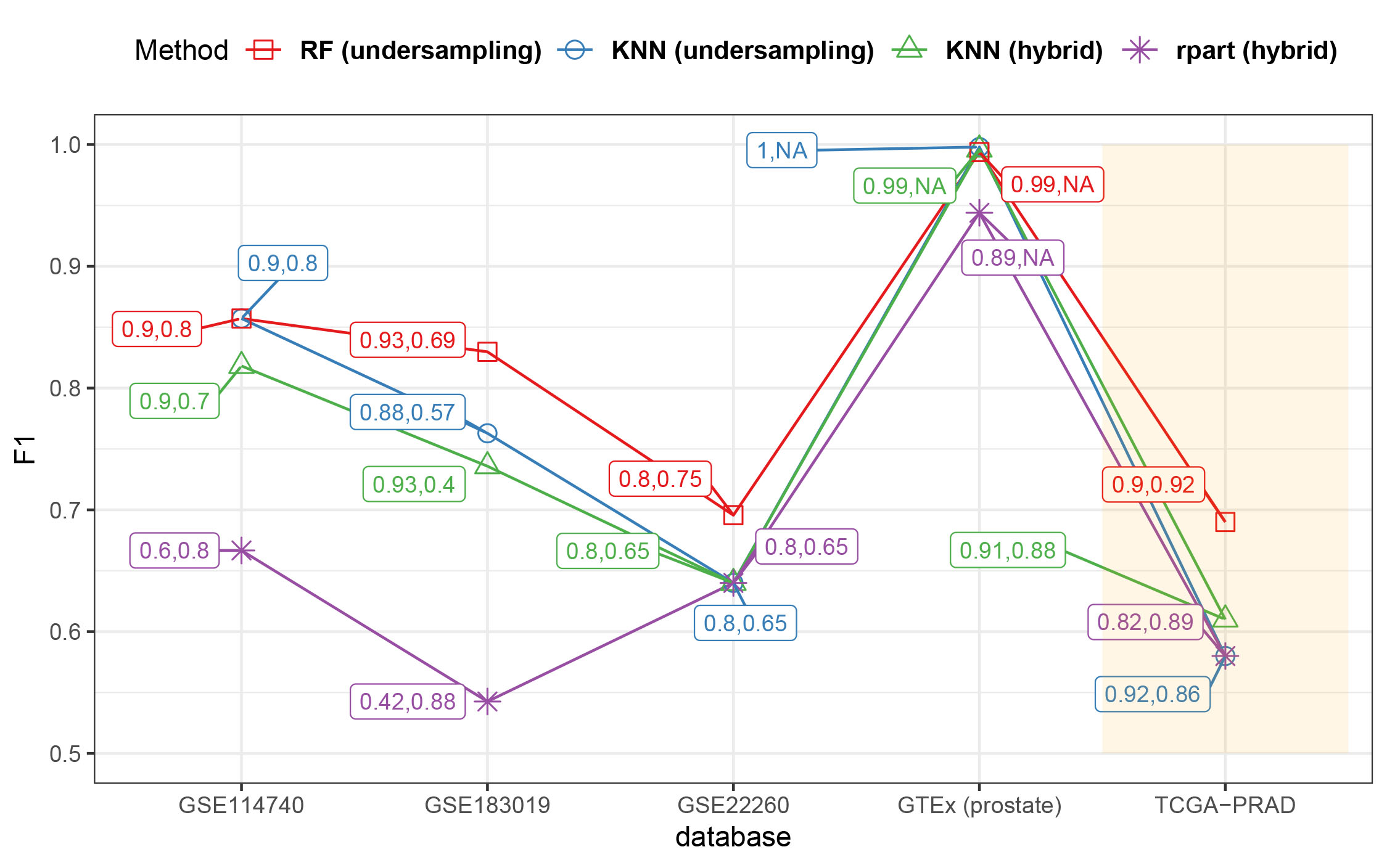
Although RF is statistically proven in our article to be performing better than the other methods, in Fig. S3 we compare our final classifier with other classifiers built in this study across external populations. Only algorithms achieving at more than 0.4 in both sensibility and specificity are represented for simplicity. This graph also proves that our classifier based on RF also works better when it comes to generalizing the results in the validation populations.
Explainability (SHAP)
Understanding why a model makes a certain prediction can be as crucial as the prediction's accuracy in many applications. However, the highest accuracy for large modern datasets is often achieved by complex models that even experts struggle to interpret, such as ensemble or deep learning models, creating a tension between accuracy and interpretability. In response, various methods have recently been proposed to help users interpret the predictions of complex models, but it is often unclear how these methods are related and when one method is preferable over another. To address this problem, SHAP (SHapley Additive exPlanations) assigns each feature an importance value for a particular prediction. SHAP, is a game theoretic approach to explain the output of any machine learning model. It connects optimal credit allocation with local explanations using the classic Shapley values from game theory and their related extensions. Shapley values are approximating using Kernel SHAP, which uses a weighting kernel for the approximation, and DeepSHAP, which uses DeepLift to approximate them.
The table below shows the 47-PCa-Genes geneset ordered according to their importance in our final classifier for the discovery population (TCGA-PRAD) as well as the validation populations.
| TCGA-PRAD | GSE114740 | GTEX | GSE22260 | GSE183019 |
| DLX1 | DLX1 | FGFR2 | DLX1 | DLX1 |
| MYL9 | HPN | CA14 | TDRD1 | TDRD1 |
| HPN | FGFR2 | ERG | MYL9 | ERG |
| FGFR2 | AMACR | HOXC6 | ERG | AMACR |
| AMACR | ANXA2 | DLX1 | HPN | HOXC6 |
| ANXA2 | HOXC6 | MYLK | ANXA2 | CAV2 |
| CNN1 | CAV2 | ANXA2 | CNN1 | FGFR2 |
| HOXC6 | CA14 | HPN | FGFR2 | MYLK |
| TDRD1 | MYLK | CAV1 | MYLK | MMP9 |
| CA14 | TDRD1 | MYL9 | EPHA2 | CXCR2 |
| MYLK | CNN1 | CAV2 | CA14 | BMP7 |
| EPHA2 | MYL9 | CNN1 | CAV2 | CA14 |
| ERG | CAV1 | DUOXA2 | PLA2G7 | CAV1 |
| CAV2 | ACTA2 | SORBS1 | AMACR | PTGS1 |
| ACTA2 | DUOXA2 | MYOCD | HOXC6 | DUOX2 |
| CAV1 | ACTG2 | BMP7 | ACTA2 | EPHA2 |
| ACTG2 | DUOX2 | MMP9 | CAV1 | AOX1 |
| DUOXA2 | PLA2G7 | ACTA2 | ACTG2 | RNF112 |
| PLA2G7 | CXCL13 | DUOX2 | CXCR2 | HPN |
| CXCR2 | ERG | CXCR2 | FLNA | SORBS1 |
| DUOX2 | SORBS1 | AMACR | DUOX2 | OR51E2 |
| BMP7 | MYOCD | PTGS1 | MMP9 | PLA2G7 |
| APOF | EPHA2 | ACTG2 | DUOXA2 | TGFBR3 |
| SORBS1 | CXCR2 | AOX1 | APOF | MYOCD |
| PTGS1 | MMP9 | EPHA2 | PTGS1 | BMP5 |
| MYOCD | GLYATL1 | TDRD1 | MYOCD | APOF |
| AOX1 | AOX1 | FLNA | AOX1 | SLPI |
| CXCL13 | RNF112 | KCNMA1 | DCN | TIMP3 |
| MMP9 | BMP7 | BMP5 | BMP7 | CXCL13 |
| FLNA | PTGS1 | SLPI | GLYATL1 | KCNMA1 |
| GLYATL1 | FLNA | LMOD1 | OR51E2 | GLYATL1 |
| MYO6 | OR51E2 | CXCL13 | MYO6 | LMO3 |
| RNF112 | APOF | OR51E2 | RNF112 | DCN |
| OR51E2 | SLPI | LMO3 | SORBS1 | LMOD1 |
| LMO3 | BMP5 | DCN | SLPI | DKK1 |
| DCN | LMOD1 | KRT7 | TIMP3 | FLNA |
| SLPI | SRARP | TGFBR3 | SVIL | MYO6 |
| BMP5 | TIMP3 | PLA2G7 | TGFBR3 | ACTA2 |
| LMOD1 | DCN | DKK1 | LMOD1 | SRARP |
| TFF3 | TFF3 | SVIL | KRT7 | SVIL |
| SRARP | KCNMA1 | SRARP | CXCL13 | DUOXA2 |
| DKK1 | KRT7 | GLYATL1 | LMO3 | ACTG2 |
| TGFBR3 | TGFBR3 | APOF | KCNMA1 | MYL9 |
| TIMP3 | MYO6 | TFF3 | DKK1 | TFF3 |
| KCNMA1 | SVIL | TIMP3 | SRARP | CNN1 |
| KRT7 | DKK1 | RNF112 | TFF3 | KRT7 |
| SVIL | LMO3 | MYO6 | BMP5 | ANXA2 |
Table S5. Gene list ordered by their SHAP importance in both the discovery and validation databases employed in this study.
The following table shows, for our discovery population (TCGA-PRAD) the mean, mininum, maximum and median contributions of each gene, ranked according to their importance. These values have been calculated for PCa and non-PCa affected tissue.This table can also be downloaded below.
| PCa Samples | Non-PCa Samples | |||||||
| Gene | mean | min | max | median | mean | min | max | median |
| DLX1 | 0.011778215 | -0.18834306 | 0.051217236 | 0.028069918 | -0.11285446 | -0.18039338 | 0.02503445 | -0.11365487 |
| MYL9 | 0.003405353 | -0.096601159 | 0.027799917 | 0.022561986 | -0.03280477 | -0.09796419 | 0.01237949 | -0.0392856 |
| HPN | 0.007259519 | -0.114094668 | 0.029144077 | 0.017216413 | -0.06959175 | -0.11915498 | 0.00759843 | -0.07376109 |
| FGFR2 | 0.006931314 | -0.083832716 | 0.030848729 | 0.013775551 | -0.06651444 | -0.08313221 | 0.00463558 | -0.07075671 |
| AMACR | 0.004705766 | -0.061588097 | 0.026595007 | 0.012454982 | -0.04516817 | -0.06100178 | 0.01564912 | -0.04905624 |
| ANXA2 | 0.004165531 | -0.052777436 | 0.027581749 | 0.014324176 | -0.03992062 | -0.05444131 | 0.01485399 | -0.0437382 |
| CNN1 | 0.002151648 | -0.075516161 | 0.016167843 | 0.012572591 | -0.02076106 | -0.07578906 | 0.00648506 | -0.0276589 |
| HOXC6 | 0.003651317 | -0.23667884 | 0.018589557 | 0.010238897 | -0.03513275 | -0.24372208 | 0.00330939 | -0.03157688 |
| TDRD1 | 0.003811285 | -0.048512562 | 0.034475567 | 0.007691927 | -0.03648479 | -0.05339948 | 0.01328714 | -0.04115016 |
| CA14 | 0.003080876 | -0.046388984 | 0.019378479 | 0.00959142 | -0.02960909 | -0.04464676 | 0.01124202 | -0.03288827 |
| MYLK | 0.002937931 | -0.060178008 | 0.012374579 | 0.008607925 | -0.02827374 | -0.06076225 | 0.00479245 | -0.03444834 |
| EPHA2 | 0.001488831 | -0.050939058 | 0.010047041 | 0.007524256 | -0.01442748 | -0.05676751 | 0.0054576 | -0.01844374 |
| ERG | 0.002184237 | -0.026131194 | 0.046507612 | -0.002256544 | -0.02100048 | -0.02676158 | 0.01622631 | -0.02351475 |
| CAV2 | 0.002526954 | -0.053569729 | 0.009449838 | 0.006235786 | -0.02427077 | -0.05929717 | 0.00472416 | -0.02889901 |
| ACTA2 | 0.000810101 | -0.028033342 | 0.007105843 | 0.005297935 | -0.00782218 | -0.02989545 | 0.00313553 | -0.00962327 |
| CAV1 | 0.001394973 | -0.031097584 | 0.006318849 | 0.00402925 | -0.01353425 | -0.02490778 | 0.00352708 | -0.01659253 |
| ACTG2 | 0.000902448 | -0.033935613 | 0.005390711 | 0.003963885 | -0.00881315 | -0.02799518 | 0.00252749 | -0.00962591 |
| DUOXA2 | 0.00110376 | -0.024988715 | 0.006139428 | 0.003684685 | -0.01063094 | -0.02319768 | 0.00273113 | -0.01351562 |
| PLA2G7 | 0.001466795 | -0.015923503 | 0.016550128 | 0.001919839 | -0.01410544 | -0.01800751 | 0.00138134 | -0.01518462 |
| CXCR2 | 0.000672123 | -0.053461734 | 0.006305264 | 0.003331705 | -0.0065194 | -0.05812444 | 0.0018352 | -0.00540055 |
| DUOX2 | 0.000717908 | -0.024372124 | 0.005498083 | 0.002799621 | -0.00700746 | -0.01968198 | 0.00347374 | -0.00803617 |
| BMP7 | 0.000478544 | -0.009723111 | 0.007322643 | 0.001463628 | -0.00465358 | -0.01268424 | 0.00440172 | -0.00521758 |
| APOF | 0.000736536 | -0.008158401 | 0.008826793 | 0.001773183 | -0.00670625 | -0.00979679 | 0.00212469 | -0.00719724 |
| SORBS1 | 0.000570459 | -0.008605752 | 0.006066128 | 0.001856945 | -0.00553485 | -0.00880035 | 0.00420633 | -0.00598885 |
| PTGS1 | 0.000585859 | -0.010262009 | 0.009097747 | 0.001376935 | -0.00557152 | -0.01587967 | 0 | -0.00619736 |
| MYOCD | 0.000546575 | -0.013096878 | 0.003827929 | 0.0015836 | -0.00530768 | -0.0119084 | 0.00204495 | -0.00573034 |
| AOX1 | 0.000757319 | -0.008875757 | 0.006705057 | 0.001017428 | -0.00722751 | -0.01097843 | 0 | -0.00779665 |
| CXCL13 | 0.000346305 | -0.035096883 | 0.004108984 | 0.001043004 | -0.00344749 | -0.04220454 | 0.0015211 | -0.0004599 |
| MMP9 | 0.00040529 | -0.020132886 | 0.011076826 | 0.000814091 | -0.00409374 | -0.01596863 | 0.00391893 | -0.00108609 |
| FLNA | 0.000351236 | -0.015885375 | 0.003348364 | 0.001107803 | -0.00361133 | -0.01667815 | 0.00112758 | -0.00410447 |
| GLYATL1 | 0.000458898 | -0.006818333 | 0.006750384 | 0.000616298 | -0.00451729 | -0.00716805 | 0 | -0.00500164 |
| MYO6 | 9.98126E-05 | -0.015081716 | 0.00606542 | 0.001158612 | -0.00104754 | -0.01798227 | 0 | -0.00071815 |
| RNF112 | 0.000164599 | -0.010876576 | 0.002949793 | 0.001150667 | -0.00197631 | -0.01336057 | 0.00128891 | -0.00183145 |
| OR51E2 | 0.000289403 | -0.009713687 | 0.004541732 | 0.000770868 | -0.00293535 | -0.00773122 | 0.00180103 | -0.00244609 |
| LMO3 | -1.79848E-05 | -0.013372757 | 0.006445573 | 0.0006329 | -0.00027267 | -0.01781317 | 0.00247417 | 0 |
| DCN | -1.42123E-05 | -0.003390791 | 0.01229765 | -0.000720507 | 0.00055049 | -0.00369968 | 0.00432751 | 0 |
| SLPI | 0.000197017 | -0.004268366 | 0.010945628 | 0 | -0.00120846 | -0.00483985 | 0.00420166 | -0.00136388 |
| BMP5 | 0.00012252 | -0.007252777 | 0.002579693 | 0.000532582 | -0.00139682 | -0.00999493 | 0 | -0.00059456 |
| LMOD1 | 0.000195481 | -0.004199646 | 0.002517822 | 0.000314099 | -0.0018711 | -0.00409386 | 0.00165005 | -0.00211689 |
| TFF3 | 0.000168114 | -0.003889075 | 0.00446708 | 0.000179987 | -0.00103796 | -0.0035612 | 0.00170565 | -0.00100177 |
| SRARP | 0.00023799 | -0.003180401 | 0.007154888 | 0 | -0.0011304 | -0.00311362 | 0.00048296 | -0.00103882 |
| DKK1 | 0.000119103 | -0.005057037 | 0.002977967 | 0 | -0.00085139 | -0.00389534 | 0.00116086 | -0.00074197 |
| TGFBR3 | 0.000101139 | -0.003295479 | 0.005980172 | 0 | -0.00071655 | -0.00292809 | 0.0020962 | -0.00017986 |
| TIMP3 | 4.25143E-05 | -0.001875877 | 0.00512233 | 0 | -0.00010633 | -0.00295412 | 0.00324269 | 0 |
| KCNMA1 | 0.000164982 | -0.005081394 | 0.002022829 | 0.00024565 | -0.00145586 | -0.00342163 | 0.00079111 | -0.00141149 |
| KRT7 | 6.93583E-05 | -0.005693768 | 0.002200788 | 0.000283169 | -0.00076019 | -0.0044584 | 0 | -0.00035849 |
| SVIL | 9.17983E-05 | -0.004864896 | 0.003586986 | 0 | -0.00053702 | -0.00341149 | 0.00113181 | -0.00025456 |
Table S6. Summary of gene contributions to the classifier output for PCa and Non-PCa samples. Mean, minimum, maximum and median values are shown for each sample group in the discovery population.
Different SHAP attribution graphs are shown in Fig. S4 for the complete set of genes in the classifier for the TCGA-PRAD database. Every dot represents a gene for a specific sample and its color is related to its expression level (red=high; blue=low).
 |
 |
 |
| Fig. S4a. Shap attribution for every gene in the full set of samples included in the TCGA-PRAD database. | Fig. S4b. Shap attribution for every gene included in the T samples for the TCGA-PRAD database. | Fig. S4c. Shap attribution for every gene included in the NT samples for the TCGA-PRAD database. |
Two different boxplots are shown in Fig. S5. On the left, the boxplot for Shap attribution is displayed for both T and NT samples separately. Similarly, on the right boxplot for gene expression is represented for T and NT samples (gene expression values have been previously normalized).
 |
 |
| Fig. S5a. Shap attribution boxplot for TCGA. | Fig. S5b. Gene expression boxplot for TCGA. |
References
- C. Genomes Project et al., "A global reference for human genetic variation," Nature, vol 526, pp. 68-74, Sep. 2015.
- F.D. Carmona et al., "A large-scale genetic analysis reveals a strong contribution of the HLA class II region to giant cell arteritis susceptibility," Am J Hum Genet, vol 96, pp. 565-580, Apr. 2015.
- H. Li and R. Durbin, "Fast and accurate short read alignment with Burrows-Wheeler transform," Bioinformatics, vol 25, pp. 1754-1760, Jul. 2009.
- B. Institute, Picard Tools, http://broadinstitute.github.io/picard/, 2018.
- A. McKenna et al., "The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data," Genome Res, vol 20, pp. 1297-1303, Sep. 2010.
- A.R. Quinlan and I.M. Hall, "BEDTools: a flexible suite of utilities for comparing genomic features," Bioinformatics, vol 26, pp. 841-842, Mar. 2010.
- S. Purcell et al., "PLINK: a tool set for whole-genome association and population-based linkage analyses," Am J Hum Genet, vol 81, pp. 559-575, Sep. 2007.
- T. K. Ho, "Random decision forests," in Proceedings of 3rd international conference on document analysis and recognition, vol. 1. pp. 278–282, 1995.
- Chen T, Guestrin C. XGBoost: A Scalable Tree Boosting System. In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining [Internet]. p. 785–94, 2016.
- N. S. Altman, "An introduction to kernel and nearest-neighbor nonparametric regression," The American Statistician, vol. 46, no. 3, pp. 175–185, 1992.
- T. Therneau and B. Atkinson, "rpart: Recursive Partitioning and Regression Trees," r package version 4.1-15, [Online]. Available: https://CRAN.R-project.org/package=rpart, 2019.