Navegating through networks.
Four cases of study:
|
>>CIS REGULATORY NETWORKS
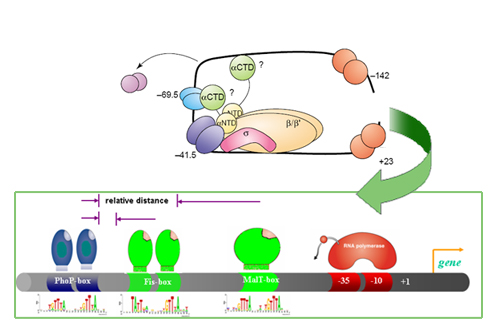
Case of study: the PhoP/PhoQ two-component regulatory system.
Problem. One of the big challenges of the post genomic era is determining when, where and for how long genes are turned on or off. Gene expression is produced by protein-protein interactions among regulatory proteins and with RNA polymerase(s), and protein-DNA interactions of these trans-acting factors with cis-acting DNA sequences in the promoters of regulated genes.
Approach. We studied the cis-features that characterize promoters controlled by the PhoP/PhoQ two-component regulatory network of Escherichia coli and Salmonella enterica serovar Typhimurium. This system is an excellent test case because it controls expression of a large number of genes in several members of the family Enterobacteriaceae, amounting to 2-3% of the genome in the case of S. enterica. Our approach is specifically designed to account for the variability in sequence, location and topology intrinsic to differential gene expression. Promoter features are analyzed concurrently, and recurrent relations are recognized to generate profiles, which are groups of promoters sharing common features. These profiles may share underlying biological properties.
Results. We uncovered novel members of, as well as regulatory interactions in the network controlled by the PhoP protein that were not discovered using previous approaches. These predictions were experimentally validated and contrasted with kinetic patterns of gene behaviors.
>> EXPRESSION NETWORKS
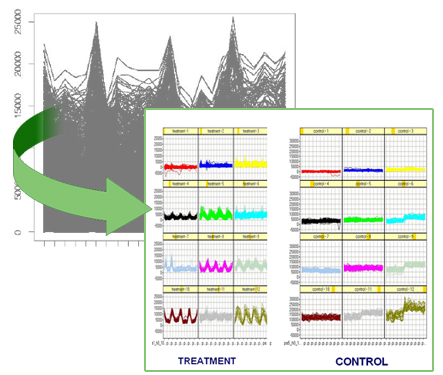
Case of study: the inflammatory response system.
Problem: Microarray technology has revolutionized modern biological research by its capacity of monitoring the expression level of thousands of genes simultaneously. However, the small number of available replicates and samples and the difficulty to identify measurement errors produced by the experimental conditions constitute the mayor drawback of this technology. A large number of new methods have rapidly emerged to provide statistical solutions to handle microarray analysis. However, there is a dearth of computational methods intended to facilitate the understanding of differential gene expression profiles (e.g., profiles that change over time and/or over treatment and/or over patient), to establish comparisons among them, and to decide which is the most reliable method to identify such profiles from collections of microarrays.
Approach. We evaluated several microarray analysis methods and found that none of these methods alone identifies all observable differential profiles that change over time and/or over treatment and/or and over subject phenotype. Consequently, we propose a method that combines the abilities of microarray data analysis methods, identifying their most representative properties, and provides a decision making set of rules that advices for their most appropriate form of application.
Results. We studied a problem derived from longitudinal blood expression profiles of human volunteers treated with intravenous endotoxin compared to placebo, as part of a Large-scale Collaborative Research Project sponsored by the National Institute of General Medical Sciences (www.gluegrant.org). Intravenous endotoxin challenge in normal volunteers is a well-characterized stimulus that reproducibly induces flu-like symptoms that resolve by 24 hours. These symptoms are associated in the model with significant changes in circulating leukocyte gene expression profiles. We identified differential profiles that where not detected by individual methods, some of which contain genes directly related to the inflammatory process. Moreover, our representation of the profiles allowed to examine the behavior of the genes independently in each subject, helping to discover the influence of biological conditions not previously considered in the experiment such as gender or age.
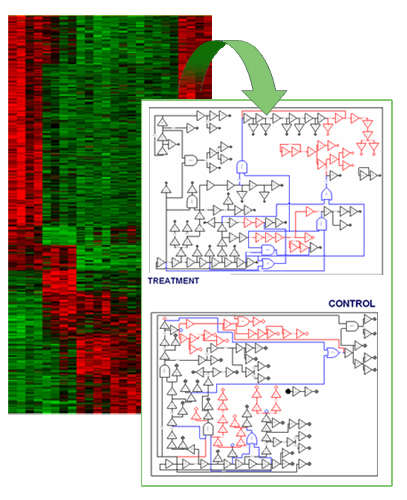
Case of study: the nerve growth factor family system.
Problem. Current developments in microarray technology have enabled a shift in the way that gene interactions can be considered namely from the simple clustering of expressions to the more complex dynamic modeling approach. Clustering techniques provide us with co-expressed groups of genes and their functional activities or co-regulated profiles. However, more informative associations can be obtained from microarray time-series. The dynamic approach assumes that gene activity is the result of a combined action of genes rather than that it is influenced by a single gene. Thus, genetic network modeling provides a methodology to employ time-series to construct a dynamic model that describes the observed phenotypic behavior based on the result of interactions between the genes and their interactions with the environment.
Approach. We propose a hierarchical learning methodology for discovering genetic networks based on large amounts of gene expression data. We build Boolean networks by developing an efficient reverse engineering algorithm. Then, we designed a conceptual clustering method, which compares different sub-networks (e.g., derived from different treatments), identifies divergent subnets, and selects only those genes included in such modules to build more detailed continuous models.
Results. We tested the ability of the proposed method to differentiate the dynamic behavior of members of the nerve growth factor (NGF) and glial cell line-derived neurotrophic factor (GDNF) families, comprising neurotrophins and GDNF-family ligands (GFLs), respectively. These factors are crucial for the development and maintenance of distinct sets of central and peripheral neurons that could be degenerates in Parkinson disease. Our approach allowed to identify relationships among genes that were undetected under typical clustering of gene expression data. This new groups are being subject of further experimental analysis.
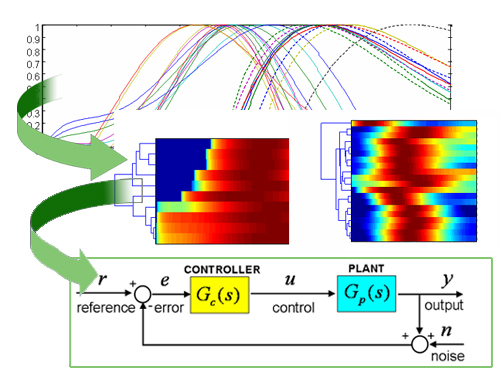
Case of study: the PhoP/PhoQ and PmrA/PmrB two-component systems.
Problem. One of the biggest challenges in genomics is the elucidation of the design principles controlling gene expression networks. Current approaches examine promoter sequences for particular features, such as the presence of binding sites for a transcriptional regulator, and identify recurrent relationships among these features termed network motifs. However, knowing the connectivity of a given network is not sufficient to define the expression dynamics of a group of genes; one also needs to specify the strength of the connections in a network or kinetic parameters, which are determined by the cis-promoter features participating in the regulation.
Approach. We identify the kinetic parameters of regulatory networks by combining experimental knowledge from binding, transcription and translation data (e.g., ChiP, quantitative PCR and GFP measurements) into mathematical models of gene expression. These models are mapped into regulatory profiles developed by identifying cis-regulatory features. The integration of this diverse knowledge (e.g., data from sequence and levels of concentration) into a common framework allows to predict gene expression from available cis-features extracted from genome sequence analysis.
Results. We studied the the PhoP/PhoQ and PmrA/PmrB two-component regulatory networks of Salmonella enterica serovar Typhimurium. The mathematical models built on the observed dynamics of transcription factor occupancy and gene transcription in vivo, assigned effective parameters determining temporal PhoP and PmrA binding and transcription to its target promoters. For example, correlation of kinetic parameters revealed that the earlier and the higher level of PhoP occupancy resulted in the faster and the stronger promoter activity of the PhoP-regulated genes. The kinetic parameters could be explained by the promoter cis-features such as the orientation and distance of the PhoP box to the RNA polymerase site and the presence of distinct PhoP box submotifs. These results were validated experimentally.
>> PATHWAY NETWORKS: GENE ONTOLOGY, PROTEIN-PROTEIN INTERACTIONS, METABOLIC PATHWAYS
Case of study: ontologies and pathway databases.
Problem. Considerable ongoing efforts are uncovering the properties of knowledge networks containing structural data including spatial maps, temporal patterns, or metabolic pathways. However, it is becoming increasingly clear that many of current efforts have been based on computational convenience of database implementers and their tendency to increase the amount of stored data. Indeed, it is often the case that available tools and techniques devoted to examine the content these large databases are hampered by their inability to support searches based on criteria that are meaningful to the users of those repositories. For example, retrieving not only tons but also known or irrelevant information. These shortcomings are particularly evident in biological networks, where clustering method originally designed for linear feature-value data are often applied. However, these databases enclose structural data that not only contains descriptions of individual observations, but also relationships among these observations. Therefore, mining into structural databases entails addressing both the uncertainty of which observations should be placed together, and also which distinct relationships among features best characterize different sets of observations.
Approach. We have addressed this problem by creating a conceptual clustering methodology that retrieves optimal descriptions of objects and interactions of objects from these network databases. Our methodology uses evolutionary computation, machine learning and multi-objective optimization techniques to generate potential hypothesis about target objects, which are described at different levels of detail and from different angles. Thus, uncovering novel solutions without being exhaustive.
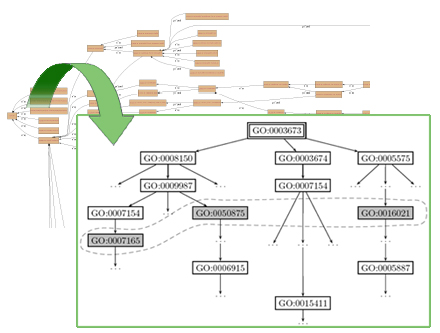
Results. We apply our methodology to the Gene Ontology database and produce annotations that can explain and predict gene expression classes derived from the analysis of longitudinal blood profiles of human volunteers in the immuno-inflammatory response problem. Analysis of the set of gene expression profiles obtained from this experiment is complex, given the number of samples taken and variance due to treatment, time, and subject phenotype. Our annotations include conjunctions of biological processes, molecular functions and cellular components defined at different levels of detail, which provide a flexible framework that describe the former complex profiles. Indeed, our approach provides resources for the annotation of groups of genes and their interactions characterized by any structural information (e.g., pathway databases).
>> FUNCTIONAL GENOMIC NETWORKS: FUSIONING GENOMIC, PROTEOMIC, AND METABOLOMIC INFORMATION
Problem
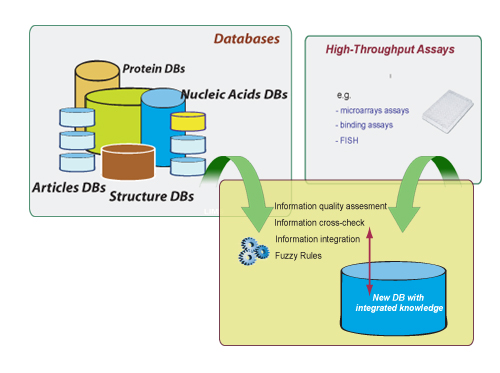
Modern molecular methods in biology and medicine are producing an unprecedented amount of data that have become an essential pool of knowledge for researchers throughout the world. This situation makes necessary the development of more sophisticated and automated ways for navigating, searching, and exploiting these data. The data in these repositories is very heterogeneous in origin, quality and annotation. Moreover, is also necessary to combine this information with those coming from different experimental technologies (DNA chips, proteomic analyses, binding, growth, mass spectrometry, FISH, PCR, Inmuno assays, cellular localization, analysis).
These technologies measure different aspects of a system differing in depth and breadth including systematic biases of a different nature. Additionally, there is usually no curated data set from which one might estimate data integration parameters, making necessary a consistent treatment of the uncertainty and false-positive and false-negative rates.
Approach. To address these concerns, we integrate, combine and generate new data depending on their quality, statistical power, type, and size. This is achieved using data mining techniques, automatically selecting through decision trees, based on previous statistical analysis, the type of algorithms or methods to be used. This approach has been successfully applied to the high throughput analysis of proteins by integration of bioinformatics and experimental data (C. del Val et al.,), and in the LIFEDB.
|


